Transthyretin amyloid cardiomyopathy (ATTR-CM) is a disease resulting from the abnormal deposition of amyloid proteins into the extracellular matrix of the myocardium. Recent data indicates that it is an underrecognised contributor to heart failure in older people.
It has classically been difficult to diagnose due to non-specific clinical manifestations, poor clinician awareness and the requirement of endomyocardial biopsy to confirm the diagnosis. Previously thought to be untreatable, there are now TTR-specific disease modifying therapies available that can slow progression and reduce mortality.
ATTR-CM is a subtype of systemic amyloidosis and is characterised by the deposition of misfolded transthyretin (TTR) protein within the myocardium. There are two distinct subtypes of ATTR-CM:
Hereditary ATTR (hATTR), resulting from mutations in the TTR gene
Wild-type ATTR (wtATTR), which is more prevalent and arises from age-related changes in the wild-type TTR protein.
While the precise prevalence of wtATTR remains unknown, it has been shown to account for up to 10% of heart failure in older patients.1 ATTR-CM can progress to an advanced stage of disease with only subtle clinical manifestations.
Non-specific symptoms combined with insufficient awareness and the absence of effective diagnostic methods have meant that this condition frequently went undetected in clinical practice. However, advances in diagnostics, with cheap and available bone-avid radiotracer scintigraphy technology, have simplified the diagnosis of this condition.2 This is allowing clinicians to make this diagnosis earlier in the disease course. The emergence of novel therapeutic options,3,4 combined with increased awareness and earlier diagnosis, has the potential to improve the prognosis of ATTR-CM.
In this case series, we describe two cases of ATTR-CM which illustrate the usual presentation of this condition, the improvement in diagnostic modalities and the current treatment options available.
Case 1 involves a 70-year-old woman who had multiple presentations to hospital with typical features of this disorder. There was a delay to diagnosis of over 18 months. In contrast, case 2 centres on a 74-year-old man who was quickly assessed as having characteristic findings of ATTR-CM and was promptly diagnosed and referred for treatment. The case discussions highlight the recent changes in diagnostic modalities and treatments available.
Case 1: A 70-year-old woman with a past medical history significant for orthostatic hypotension and polycythaemia vera (PV), presented to the emergency department (ED), with presyncope, dyspnoea on exertion and atypical chest pain
She denied any episodes of syncope but admitted to paraesthesia in the lower limbs, which had been ongoing for at least two years. The episodes of presyncope mainly occurred upon standing but could also occur intermittently when seated.
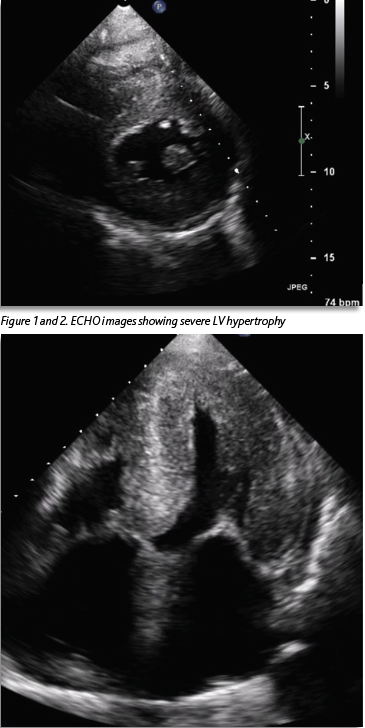
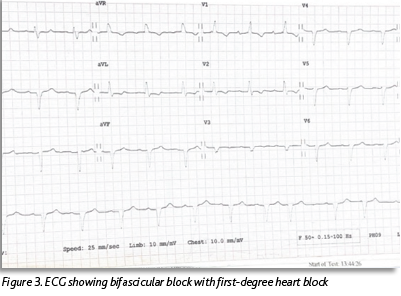
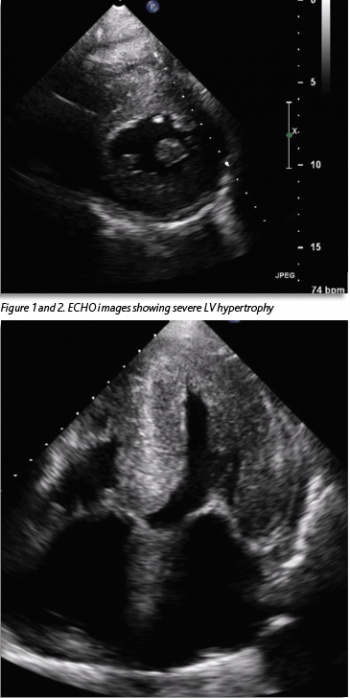
She was known to have severe left ventricular hypertrophy (LVH) with preserved LV function, which had been seen on echocardiogram (ECHO) two years previously (Figures 1 and 2). A coronary angiogram two years previously showed no evidence of coronary artery disease. Her ECG showed a new bifascicular and first-degree heart block compared to one from two years prior (see Figure 3). Troponin level was elevated at 30ng/L, which was stable from the previous admission (32ng/L). Otherwise, laboratory investigations were unremarkable. She had a significant postural drop in systolic blood pressure of 21mmHg on standing. Her medication regimen included aspirin 75mg, lansoprazole 30mg and hydroxycarbamide 500mg. There was no notable family history. She described a similar presentation to another hospital three months previously. Her vital signs were within normal limits. Cardiovascular examination was unremarkable and the patient was euvolaemic.
(click to enlarge)
(click to enlarge)
This patient was admitted on continuous cardiac monitoring and continued to have orthostatic hypotension over the course of the admission. The patient was discharged two days later with advice on maintaining adequate hydration. Follow-up was planned with the falls and syncope unit in the hospital. She re-presented to ED three months later with similar symptoms. ECG, laboratory investigations and ECHO were repeated at that stage and showed similar findings.
A stress perfusion cardiac MRI was performed at this stage. The report was significant for circumferential subendocardial hypoperfusion during vasodilator stress, rapid washout of gadolinium post-contrast with extensive subendocardial diffuse late gadolinium enhancement and late atrial wall gadolinium enhancement. This constellation of features was reported as being typical for cardiac amyloid.
At this stage the patient was referred for endomyocardial biopsy, as bone-avid radiotracer scintigraphy technology had not yet been implemented as the standard practice. The biopsy samples were sent to the UK for further analysis, further prolonging the time to a confirmed diagnosis. The team also arranged testing for AL amyloid including serum kappa/lambda free light chain ratio analysis, serum protein immunofixation and urine protein immunofixation, which was negative. The patient remained as an inpatient for five further days before the endomyocardial biopsy was performed. At this time she was started on pregabalin 75mg for peripheral paraesthesia. Neurological exam at this time revealed evidence of peripheral neuropathy. Fludrocortisone was started for symptoms related to orthostatic hypotension.
The patient was discharged home with a plan for early outpatient follow-up. The endomyocardial biopsy results confirmed ATTR-CM and genetic testing was positive for the T60A variant of hereditary amyloidosis. Further investigations were performed including neurophysiology which revealed bilateral carpal tunnel lesions and large fibre, length-dependent peripheral neuropathy. She was subsequently started on patisiran, an RNA interference agent, which can delay the progression of ATTR-CM.
As the condition progressed, LV function began to deteriorate. This was associated with the development of cardiorenal syndrome. The symptoms of postural hypotension also began to worsen requiring the addition of midodrine. Furosemide 20mg was started after the development of pedal oedema. She also developed altered bladder function which necessitated long-term catheterisation and gastrointestinal dysmotility causing diarrhoea. The patient was linked in with palliative care as she continued to deteriorate. She died three years after initial diagnosis, from acute decompensated heart failure.
Case discussion
This case illustrates very well the typical presentation of a patient with ATTR-CM and highlights both the prevailing lack of awareness regarding this condition and the difficulty faced by clinicians in diagnosing it. The patient initially exhibited a range of features commonly associated with ATTR-CM such as left ventricular hypertrophy and preserved ejection fraction on ECHO, conduction abnormalities on ECG, a mild, chronic troponin elevation and discordance between the degree of LV thickness on ECHO and QRS voltage on ECG.5
The symptoms that she presented with such as presyncope and dyspnoea, while non-specific, are commonly seen in ATTR-CM. Similarly there were features of neurological dysfunction which are commonly seen in ATTR amyloidosis, such as autonomic dysregulation resulting in orthostatic hypotension and peripheral neuropathy causing symptoms of peripheral sensory disturbance. Other features typical of ATTR amyloidosis that developed as the disease progressed included bladder dysfunction and gastrointestinal dysmotility.
There was a diagnostic delay of approximately 18 months in this case, after the first ECHO showing severe LVH was performed, with multiple presentations to hospital in that time. If left untreated, ATTR-CM with evidence of heart failure is associated with a life expectancy of only 2.5-3.5 years.6 Given the progressive nature of this condition and serious prognosis without treatment, early diagnosis is crucial.
This case also highlights the previous diagnostic challenges that were associated with this condition. The necessity for endomyocardial biopsy resulted in more time in hospital, exposed the patient to an invasive procedure and also required a prolonged time period before the diagnosis could be confirmed.
This patient was treated with patisiran, a TTR-specific therapy. This was originally approved for ATTR polyneuropathy but subgroup analysis of patients with cardiac amyloidosis enrolled in a trial of patisiran showed reduced LV wall thickness and N-terminal pro-B-type natriuretic peptide (NT-proBNP) levels and slowed the progression of LV systolic dysfunction.7 The other therapies that are available for ATTR-CM include tafamidis, which prevents cleavage of transthyretin tetramers and may reduce deposition of amyloid. This has been shown to have beneficial effects on mortality and cardiovascular hospitalisation. Tafamidis has recently been approved for reimbursement for ATTR-CM patients in Ireland.8
Management of heart failure in these patients involves general measures such as sodium and fluid restriction and alcohol avoidance. Diuretics are used as required to relieve symptoms and maintain euvolaemia. This can be difficult as ventricular capacity is markedly reduced.
There are no specific guidelines surrounding the use of neurohormonal agents; however angiotensin converting enzyme (ACE) inhibitors and angiotensin receptor blockers (ARBs) tend to be poorly tolerated due to hypotension. Vasodilator medications and those with negative chronotropic and inotropic effects such as beta blockers, should also be avoided.9
Case 2: A 74-year-old man presented to the ED with acute onset of presyncope and dyspnoea on exertion
Heart rate on presentation was 36bpm and blood pressure was 128/87mmHg. ECG showed slow atrial fibrillation with complete heart block. Past medical history was significant for bilateral carpal tunnel syndrome, ischaemic stroke, atrial fibrillation, hypertension and thymoma resection. There was no relevant family history. His medication regimen included rivaroxaban 20mg, ramipril 1.25mg, bisoprolol 2.5mg, atorvastatin 40mg and pantoprazole 40mg.
On examination, there was mild pitting oedema but otherwise the cardiovascular exam was unremarkable. Echocardiogram showed moderate LV hypertrophy and global hypokinesis with an ejection fraction of 30-40%. Laboratory investigations were all within normal reference range except for a BNP of 565ng/L (ref < 100).
The patient was admitted on telemetry. Bisoprolol was withheld and the patient’s heart rate remained stable without inotropic support. He was referred to a tertiary centre for pacemaker insertion. A single-lead pacemaker was inserted without complication on day seven of admission. The cardiologist who implanted the pacemaker identified multiple features consistent with ATTR-CM, including conduction disease, LVH and LV dysfunction on ECHO and bilateral carpal tunnel syndrome, and so advised cardiac MRI.
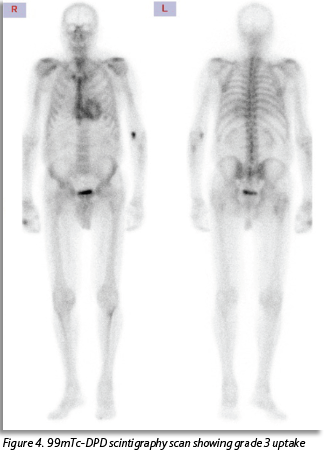
MRI showed rapid washout of gadolinium post-contrast with subendocardial late gadolinium enhancement suggestive of amyloidosis. Instead of being referred for endomyocardial biopsy this patient underwent a 99mTc-DPD scintigraphy scan. This revealed radioisotope uptake consistent with amyloidosis (see Figure 4).
(click to enlarge)
Tests for AL amyloid were negative, as were genetic tests for hereditary ATTR, leading to a diagnosis of wild-type ATTR (wtATTR). The patient was referred to an amyloidosis clinic for further management and is currently being considered for amyloid-specific therapies. He is currently stable from a heart failure standpoint.
Case discussion
This case serves as a noteworthy illustration of the growing recognition of ATTR-CM in recent years. This patient had numerous features in the history, concerning for ATTR-CM, including conduction disease on his ECG, LV hypertrophy and dysfunction seen on ECHO, and a previous diagnosis of atrial fibrillation which is commonly seen in these patients. Furthermore, his medical history was significant for bilateral carpal tunnel syndrome, a recognised early indicator of amyloidosis.10 The cardiologist in this case was able to recognise this constellation of conditions and the associated risk of underlying amyloidosis. This underlies the importance of increasing the awareness of this condition within the medical community.
This case also illustrates the improvement in diagnostic modalities for ATTR-CM. Bone-avid radiotracer scintigraphy technology in the form of a 99mTc-DPD scan, is a non-invasive and readily available imaging modality. Patients who test negative for AL amyloid can avoid an endomyocardial biopsy. In the absence of an abnormal serum free light chain ratio or detectable monoclonal proteins, the specificity of bone scintigraphy for ATTR-CM is 100%.11 The sensitivity of the DPD scan for detecting amyloid deposits is > 99%.11 The timely diagnosis of disease in this case was crucial, given the potential utility of disease-modifying TTR-specific therapies. Timely and accurate diagnosis also ensures tailored management of heart failure, avoiding poorly tolerated treatments like vasodilators or negative inotropic/chronotropic agents. Additionally, it allows clinicians to be vigilant for other complications of amyloidosis that may arise.
Conclusion
Enhanced diagnostic methods have led to greater recognition of ATTR-CM, a condition once marred by diagnostic delays and poor outcomes. The above two contrasting cases underscore the critical significance of early detection and diagnosis, facilitating timely referral for disease-modifying interventions.
These cases succinctly illustrate the challenges present in recognising, diagnosing and managing patients with ATTR-CM. Recent data suggests that the rate of ATTR-CM in older patients with heart failure is far higher than previously thought, especially in those with heart failure with preserved ejection fraction, low flow aortic stenosis and increased ventricular wall thickness.12 As a result, this condition has transitioned from being a seldom-seen and poorly understood disorder, once considered as progressive and untreatable, to one that is increasingly recognised in clinical practice.
Case 1 underlies the diagnostic delay frequently present in patients with ATTR-CM, due to poor recognition of the typical presentation. This leads to a delay in instigating the appropriate treatment strategies and allows patients to reach an advanced stage of disease before receiving an accurate diagnosis.
Increased awareness of the disease and its characteristic features combined with an easily accessible and non-invasive diagnostic modality has resulted in more patients receiving an accurate and timely diagnosis earlier in the disease course. These individuals have the potential to benefit from recently approved disease-modifying therapies, which can significantly improve long-term survival and reduce cardiovascular hospital admissions.
References
Witteles R, Bokhari S, Damy T et al. Screening for transthyretin amyloid cardiomyopathy in everyday practice. J Am Coll Cardiol HF 2019 Aug 7; (8):709-16
Hanna M, Ruberg FL, Maurer MS et al. Cardiac scintigraphy with technetium-99m-labeled bone-seeking tracers for suspected amyloidosis: JACC Review Topic of the Week. J Am Coll Cardiol 2020 Jun 9; 75(22):2851-62. doi: 10.1016/j.jacc.2020.04.022
Adams D, Gonzalez-Duarte A, O’Riordan WD et al. Patisiran, an RNAi therapeutic, for hereditary transthyretin amyloidosis. N Engl J Med 2018; 379:11-21
Benson MD, Waddington-Cruz M, Berk JL et al. Inotersen treatment for patients with hereditary transthyretin amyloidosis. N Engl J Med 2018; 379:22-31
Pagura L, Porcari A, Cameli M, et al. ECG/echo indexes in the diagnostic approach to amyloid cardiomyopathy: A head-to-head comparison from the AC-TIVE study. Eur J Intern Med 2023, Oct 17. doi:10.1016/j.ejim.2023.09.026
Gonzalez-Lopez E, Escobar-Lopez L, Obici L et al. Prognosis of Transthyretin Cardiac Amyloidosis Without Heart Failure Symptoms. JACC CardioOncol 2022, Nov 15; 4(4):442-54. doi: 10.1016/j.jaccao.2022.07.007
Adams D, Gonzalez-Duarte A, O’Riordan WD et al. Patisiran, an RNAi Therapeutic, for Hereditary Transthyretin Amyloidosis. N Engl J Med 2018; 379(1):11-21. doi:10.1056/NEJMoa1716153
Maurer MS, Schwartz JH, Gundapaneni B et al. Tafamidis Treatment for Patients with Transthyretin Amyloid Cardiomyopathy. N Engl J Med 2018; 379(11):1007-16. doi:10.1056/NEJMoa1805689
Ruberg FL, Grogan M, Hanna M, Kelly JW, Maurer MS. Transthyretin amyloid cardiomyopathy: JACC State-of-the-Art Review. J Am Coll Cardiol 2019, Jun 11; 73(22):2872-91. doi: 10.1016/j.jacc.2019.04.003
Nakagawa M, Sekijima Y, Yazaki M et al. Carpal tunnel syndrome: a common initial symptom of systemic wild-type ATTR (ATTRwt) amyloidosis. Amyloid 2016; 23:58
Gillmore JD, Maurer MS, Falk RH et al. Non-biopsy diagnosis of cardiac transthyretinamyloidosis. Circulation 2016; 133:2404-12
González-López E, Gallego-Delgado M, Guzzo-Merello G et al. Wild-type transthyretinamyloidosis as a cause of heart failure with pre-served ejection fraction. Eur Heart J 2015; 36:2585-94
 (click to enlarge)
(click to enlarge)